Overview: The Core Principle of Preclinical Safety in Latin America
The Regulatory Foundation: Understanding ISO 10993 and ISO 14971
A Practical Guide to Building Your Preclinical Safety Dossier
Regulatory Landscape: A Country-by-Country Analysis
Common Pitfalls in Preclinical Submissions (And How to Avoid Them)
Conclusion: Latin America's Strategic Advantage for MedTech Innovators
Frequently Asked Questions (FAQ)
Sources
Overview: The Core Principle of Preclinical Safety in Latin America
The Direct Answer: No Mandated Animal Study Checklist
Foreign medical device sponsors planning a First-in-Human (FIH) clinical trial in Latin America often ask for a checklist of required preclinical animal studies. The direct answer is that one does not exist. Regulatory agencies across the region, including Colombia's National Food and Drug Surveillance Institute (INVIMA), do not maintain a prescriptive list of animal tests that must be performed before an FIH trial application.1
This reality marks a significant departure from older, more rigid regulatory models. The focus has shifted from the means of testing to the end result: a robust and scientifically sound demonstration that the investigational device is safe for initial use in human subjects. This flexibility is not a regulatory gap but a deliberate, modern approach.
The Real Requirement: Demonstrating Safety Through a Risk-Based Approach
The fundamental requirement from agencies like INVIMA, Brazil's ANVISA, and Mexico's COFEPRIS is that sponsors provide a comprehensive safety dossier that establishes a "reasonable presumption of safety".1 This dossier is not built by ticking boxes on a testing menu but is instead the output of a thorough, device-specific risk assessment process.
Sponsors must scientifically justify their testing strategy. This involves a deep analysis of the device's materials, manufacturing processes, design, and intended clinical use—including the nature and duration of tissue contact.2 The central task is to identify all potential biological hazards and then systematically address them with the most relevant evidence, which may or may not involve new animal testing.
From Prescription to Justification: A Favorable Shift for Innovators
This regulatory environment, which prioritizes justification over prescription, is highly advantageous for innovative MedTech startups. It allows sponsors to use modern, efficient, and often more scientifically relevant methods to prove safety. This can avoid costly and time-consuming animal studies that may not be necessary for a specific device, freeing up critical capital and accelerating development timelines.3
This approach is also deeply aligned with the globally accepted ethical principles of the "3Rs" (Replace, Reduce, Refine) for animal testing.2 By requiring sponsors to first exhaust all other data sources and alternative methods, Latin American regulators are promoting a more ethical and scientifically advanced paradigm. The entire region has largely converged on a set of international standards as the foundation for this approach. The adoption of standards like ISO 10993 and ISO 14971 by countries from Mexico to Argentina creates a harmonized scientific framework.1 This harmonization means that a core preclinical data package, if built correctly, is largely portable across the region, simplifying multi-country clinical trial strategies and lowering the barrier to entry for foreign sponsors.
The Regulatory Foundation: Understanding ISO 10993 and ISO 14971
ISO 10993: The Global Standard for Biological Evaluation
The International Organization for Standardization's 10993 series, "Biological evaluation of medical devices," is the universally recognized framework for assessing a device's biocompatibility. It is the cornerstone of preclinical safety submissions in Latin America, referenced by regulators in Brazil, Argentina, Peru, Ecuador, and Paraguay, among others.1
ISO 10993 is not a simple checklist. It is a risk-based framework that guides a sponsor through a logical evaluation process. It categorizes devices based on the nature of body contact (e.g., surface, implant) and the duration of that contact (e.g., limited, prolonged, permanent). This categorization helps determine which potential biological effects (e.g., cytotoxicity, sensitization, systemic toxicity) must be evaluated for a given device.2
The 2018 Paradigm Shift: Why Chemical Characterization is Now King
A critical development that underpins the current regulatory flexibility in Latin America was the 2018 revision of the foundational document in the series, ISO 10993-1.1 This update represented a major paradigm shift in biocompatibility assessment.
Before 2018: The standard was often misinterpreted as a menu of biological tests to be performed based on device category.
After 2018: The revision clarified that the primary goal is a comprehensive biological risk assessment. It established physicochemical characterization as the essential starting point and the only universally mandatory requirement.
The modern approach, catalyzed by this ISO update, dictates that sponsors must first thoroughly understand the chemistry of their device. This involves identifying and quantifying any chemical substances that could potentially be released from the device during use (extractables and leachables). The toxicological risks of these identified chemicals are then assessed against established safety thresholds. Biological testing, particularly animal testing, is positioned as a method to be used only when this chemical and toxicological assessment cannot sufficiently address a potential risk.1 This change in the international standard provided regulatory bodies like INVIMA and ANVISA with the formal, scientifically validated justification to move away from demanding specific animal tests by default.
ISO 14971: The Role of Risk Management in Your Testing Strategy
The ISO 14971 standard, "Medical devices — Application of risk management," is inextricably linked to ISO 10993. It is the procedural backbone of the entire safety evaluation. The risk management process, which must be documented in a Risk Management File, is where a sponsor identifies the biological hazards that the biocompatibility evaluation needs to address.4
For example, a risk analysis for an implantable device would identify potential hazards like material toxicity, thrombogenicity, and effects of degradation products. The biocompatibility testing plan (or the justification for not conducting certain tests) must be a direct and logical output of this documented risk analysis. A sponsor cannot simply choose tests from a list; they must demonstrate that their evaluation plan is a tailored response to the specific risks posed by their specific device.
The 3Rs (Replace, Reduce, Refine): The Ethical Framework
The modern, risk-based approach is built on the ethical framework of the "3Rs" 2:
Replace: Replace animal testing with non-animal methods whenever possible. This includes using existing data, in vitro cell culture assays, and computer (in silico) modeling.
Reduce: Reduce the number of animals used to the minimum necessary to obtain scientifically valid results.
Refine: Refine procedures to minimize any potential pain or distress to the animals involved.
Latin American regulators have embraced this framework. Demonstrating that an animal test is not scientifically necessary is now considered a best practice. Brazil's ANVISA, for example, explicitly encourages the use of New Approach Methodologies (NAMs) as alternatives to animal testing in its regulations.1 This shows a commitment not just to international standards, but also to the ethical principles that guide modern medical research.
A Practical Guide to Building Your Preclinical Safety Dossier
To meet the expectations of Latin American regulators, sponsors should follow a systematic, evidence-based process. This approach focuses on building a scientific argument for safety rather than simply completing a list of tests.
Step 1: Conduct a Comprehensive Biological Risk Assessment (ISO 14971)
The process begins with a formal risk assessment as defined by ISO 14971. This is not a cursory exercise but a foundational document.
Identify Materials and Processes: Document every material and chemical used in the device and its manufacturing, including colorants, plasticizers, and processing aids.
Define Intended Use: Clearly define the device's clinical application, the nature of body contact (e.g., skin, blood, bone), and the duration of that contact.
Identify and Document Hazards: Systematically identify all potential biological hazards based on the device's characteristics. This could include cytotoxicity, irritation, sensitization, systemic toxicity from leached chemicals, or thrombogenicity for blood-contacting devices.
Justify Applicability: For each potential hazard, document the rationale for why it is or is not considered a relevant risk for your device.1
Step 2: Prioritize Physicochemical Characterization and Analysis
As mandated by the 2018 update to ISO 10993-1, the next step is to understand the device's chemical composition and what substances it might release.
Chemical Characterization: Perform a comprehensive chemical characterization of all patient-contacting materials.
Extractables & Leachables (E&L) Testing: Conduct studies using exaggerated conditions (e.g., aggressive solvents, high temperatures) to identify all potential "extractables." Then, conduct studies under conditions that simulate clinical use to identify "leachables"—the substances that would actually be released into the body.
Quantify and Compare: Quantify the identified leachables and compare these levels against established toxicological thresholds from scientific literature and regulatory guidance.1
Step 3: Leverage Existing Data (Literature, Predicates, and Material Data)
Before commissioning any new biological tests, sponsors must conduct a thorough search for existing data. This is a critical part of the justification process.
Systematic Literature Review: Perform and document a systematic review of scientific literature on the biocompatibility of the device's materials (e.g., medical-grade titanium, polycarbonate).
Predicate Device Data: If the device is similar to other legally marketed devices, leverage the clinical and preclinical safety data available for those "predicate" devices.
Material Supplier Data: Obtain biocompatibility data and Master Files from the suppliers of the raw materials used in the device.1
Step 4: Employ Alternative Testing Methods (In Vitro, In Silico, NAMs)
If the risk assessment and existing data are insufficient to address a particular hazard, the next step is to use alternative, non-animal methods.
In Vitro Assays: Use standardized in vitro tests, such as ISO 10993-5 for cytotoxicity, which uses cell cultures to assess toxicity.
In Silico Modeling: Use computational models to predict a chemical's toxicity based on its structure (a "read-across" approach).
New Approach Methodologies (NAMs): This broad category includes a range of advanced non-animal methods. Their use is explicitly encouraged in some countries, like Brazil, demonstrating a progressive regulatory stance.1
Step 5: Author a Robust Justification Memorandum for Your Testing Strategy
This final document is the capstone of the preclinical submission. It synthesizes all the information from the previous steps into a single, cohesive scientific argument that explains why the device is safe for FIH testing. The quality of this memorandum is paramount.
The shift from a checklist-based system to a justification-based one has elevated the importance of a specific role on the development team: the toxicologist. Previously, a lab technician could run a test and produce a pass/fail report. Now, a sponsor generates complex chemical data that requires expert interpretation. A qualified toxicologist is needed to analyze the E&L data, assess the biological risk of each identified chemical, and author the expert evaluation that forms the core of the justification memorandum. Submitting raw chemical data without this expert toxicological assessment is a common reason for rejection or significant delays.1 Therefore, startups must secure access to this expertise early in their development process.
The final justification memorandum must include:
A detailed rationale for the evaluation strategy chosen.
The expert toxicological risk assessment.
A comprehensive risk-benefit analysis for the FIH trial.
Clear documentation linking all the evidence back to the biological risks identified in Step 1, demonstrating that every potential risk has been adequately addressed.1
Regulatory Landscape: A Country-by-Country Analysis
While the scientific principles are harmonized across Latin America, each country has its own regulatory body and specific administrative procedures. However, the foundational reliance on a risk-based approach and international standards provides a consistent platform for sponsors. Regional economic blocs like the Andean Community (CAN) and MERCOSUR further drive this harmonization, meaning the regulatory language and expectations in member countries like Ecuador, Peru, Paraguay, and Argentina are often very similar.6
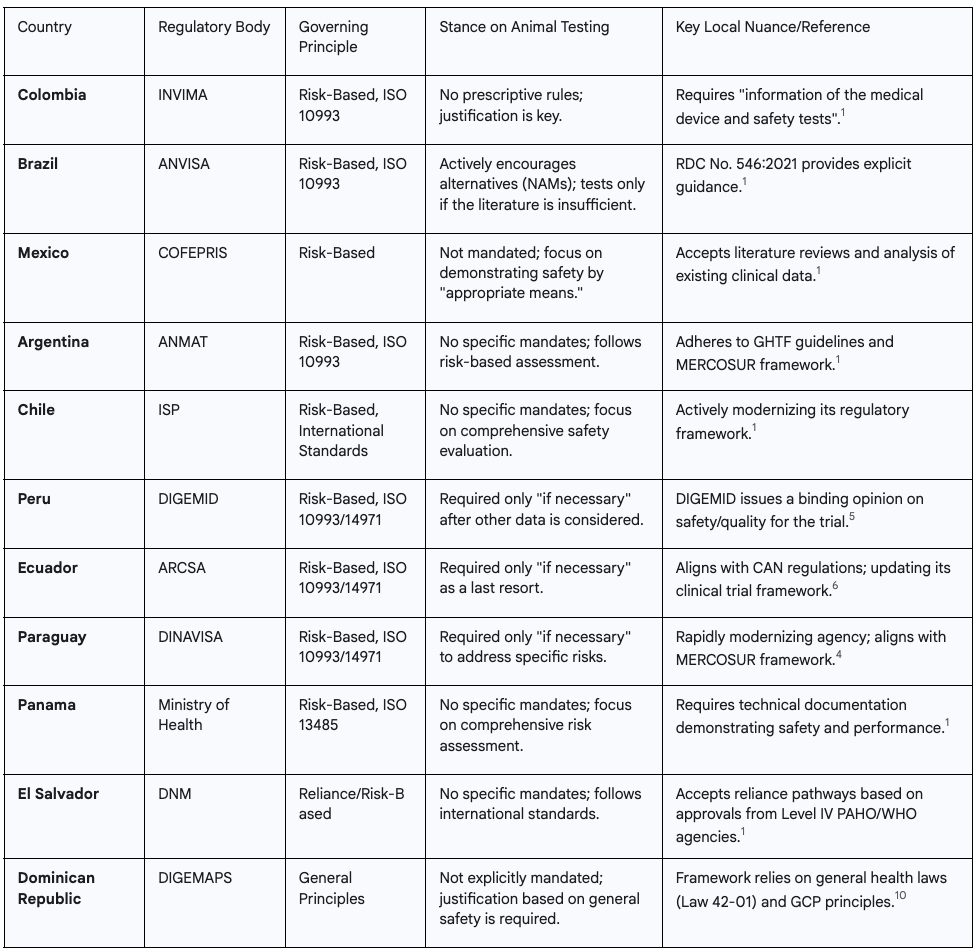
Table: Preclinical Requirements Snapshot for FIH Medical Device Trials in Latin America
Detailed Country Profiles
Colombia (INVIMA): As the primary subject of the initial query, Colombia is a prime example of the regional approach. INVIMA's regulations require the submission of "information of the medical device and safety tests" for a clinical trial application. There are no prescriptive rules dictating which tests must be animal-based. The emphasis is entirely on the sponsor providing a well-documented, scientifically sound rationale that demonstrates the device's safety for human use.
Brazil (ANVISA): Brazil is arguably the most progressive country in the region regarding alternatives to animal testing. ANVISA's Resolution RDC No. 546:2021, which governs biocompatibility, explicitly states that "specific assays must be performed only when a complete literature review is insufficient to secure biocompatibility." The framework actively encourages the use of New Approach Methodologies (NAMs), signaling a clear preference for non-animal data when scientifically appropriate.
Mexico (COFEPRIS): Mexico's regulatory authority, COFEPRIS, also follows a flexible path. For new devices, the focus is on demonstrating safety and efficacy through appropriate means, which does not necessarily mean animal testing. The agency accepts clinical evaluations that can be based on comprehensive literature reviews and analyses of available clinical data for similar devices.
Argentina (ANMAT): Argentina's National Administration of Drugs, Foods and Medical Devices (ANMAT) aligns with the MERCOSUR framework, Global Harmonization Task Force (GHTF) guidelines, and ISO 10993 standards. Like its neighbors, the emphasis is on a risk-based assessment of biocompatibility rather than a checklist of mandatory animal tests.
Chile (ISP): The Public Health Institute of Chile (ISP) is in the process of modernizing its regulatory framework but already aligns with international standards. Its focus for clinical trial approval is on a comprehensive evaluation of the device's safety and performance, without specific mandates for animal testing.
Peru (DIGEMID): Peru's framework, overseen by the General Directorate of Medicines, Supplies and Drugs (DIGEMID), explicitly requires a risk analysis according to ISO 14971 and biocompatibility evaluation according to ISO 10993. Animal studies are only required "if necessary" to assess safety and efficacy before human trials. As part of the clinical trial authorization process, DIGEMID issues a binding technical opinion on the safety and quality of the investigational product.
Ecuador (ARCSA): Ecuador's Agency for Health Regulation, Control and Surveillance (ARCSA) follows a nearly identical path to Peru, requiring risk management per ISO 14971 and biocompatibility per ISO 10993. Animal studies are considered only when necessary. The country is actively updating its clinical trial regulations with PAHO assistance to further align with the highest international ethical and regulatory standards.
Paraguay (DINAVISA): Paraguay's National Directorate of Sanitary Surveillance (DINAVISA) has undergone rapid modernization, implementing a digital submission portal and aligning with the latest MERCOSUR framework. Its technical requirements mirror those of its neighbors, demanding a risk analysis (ISO 14971), biocompatibility evaluation (ISO 10993), and only requiring animal studies "if necessary".
Panama (Ministry of Health): Panama's approach is centered on comprehensive technical documentation. Sponsors must demonstrate device safety and performance, with quality systems adhering to ISO 13485. The focus is on the overall risk assessment rather than specific preclinical test requirements.
El Salvador (DNM): El Salvador's National Directorate of Medicines (DNM) leverages the work of other respected agencies. It recognizes approvals from Level IV PAHO/WHO certified agencies (like ANVISA and COFEPRIS) and accepts reliance pathways. This can significantly expedite the approval process for devices that have already undergone rigorous review elsewhere.
Dominican Republic (DIGEMAPS): The regulatory framework in the Dominican Republic, managed by the General Directorate of Medicines, Food and Health Products (DIGEMAPS), is less explicitly defined than in other major Latin American markets. The requirements are derived from general principles within the General Health Law (No. 42-01) and an expectation of adherence to Good Clinical Practice (GCP). While preclinical investigations, including biocompatibility and toxicology assessments, are expected to establish a foundation of safety, the absence of specific regulations makes the sponsor's own robust, well-documented safety justification even more critical.
Common Pitfalls in Preclinical Submissions (And How to Avoid Them)
Navigating this flexible but rigorous environment requires avoiding common mistakes that can lead to delays or rejection.
Pitfall 1: The Superficial Literature Review
Regulators expect a systematic, scientific, and well-documented literature review, not a superficial search. A weak review is a major red flag.
How to Avoid: Conduct a structured review with a clear protocol, search terms, and inclusion/exclusion criteria. The final report should not just list papers but analytically summarize the findings and draw clear conclusions about how the existing evidence supports the safety of your device's materials and design.
Pitfall 2: Data Without Interpretation (The Missing Toxicologist)
As previously noted, one of the most common failings is submitting raw data, particularly from chemical characterization, without an expert toxicological evaluation. Regulators are not there to interpret your data for you.
How to Avoid: Engage a qualified toxicologist early. Their role is to translate the chemical data into a biological risk assessment, explaining what the presence of certain chemicals at certain levels means for patient safety. This expert report is a non-negotiable component of a successful submission.
Pitfall 3: Ignoring Local Administrative Requirements
While the scientific principles of preclinical safety are harmonized across Latin America, the administrative processes for submitting a clinical trial application are not. This contradiction—harmonized science, divergent process—is a critical trap for sponsors. A perfect scientific dossier can be rejected on a simple administrative technicality.
How to Avoid: Meticulous, country-specific preparation is essential. This includes:
Language: Ensuring all required documents are professionally translated into Spanish (or Portuguese for Brazil).
Formats: Using the correct application forms for each country, such as those required by Peru's INS.
Submission Portals: Understanding and correctly using the required digital submission systems, like Paraguay's DINAVISApy e-Portal or Ecuador's online platform.
Local Nuances: Adhering to all local requirements, which may seem minor but are mandatory. This highlights the value of working with a local expert or CRO who is familiar with these specific national procedures.
Pitfall 4: The "Assumption of Acceptance" Trap
Sponsors sometimes misinterpret "no specific requirement for animal testing" as "no requirements at all." This is a dangerous assumption. The flexibility of the system places the entire burden of proof squarely on the sponsor.
How to Avoid: Adopt the mindset that you must build an unassailable case for safety from the ground up. Every claim must be backed by evidence. The absence of a specific rule does not lower the standard of evidence required; it simply gives you more flexibility in how you generate that evidence.
Conclusion: Latin America's Strategic Advantage for MedTech Innovators
Recap: A Focus on Sound Science, Not Inflexible Rules
The preclinical regulatory landscape for medical devices in Latin America is modern, efficient, and science-driven. For a foreign startup asking about mandatory animal studies, the answer is clear: the focus is not on a prescribed list of tests but on a sponsor's ability to build a robust, scientifically-justified safety case. By harmonizing around international standards like ISO 10993 and ISO 14971, the region's regulators have created a predictable and flexible environment that rewards innovation while upholding the highest standards of patient safety.
The Business Case: Accelerating Your Path to FIH Trials
This regulatory approach is not just a procedural detail; it creates a significant strategic opportunity for MedTech innovators. For a startup, where time and capital are the most critical resources, the advantages are compelling.
Faster Approval Timelines: The pragmatic and efficient review processes can lead to FIH trial approvals in as little as 2-4 months, compared to 6-12 months or more in the U.S. or Europe.
Cost Efficiency: Trial costs can be approximately 30% lower than in the U.S. or EU. This is due in part to lower operational costs, but also to the ability to avoid expensive, unnecessary animal studies through a strong scientific justification.
High-Quality Infrastructure: The region boasts a growing ecosystem of experienced clinical research sites and regulatory authorities recognized as Level IV by PAHO/WHO, ensuring that trials are conducted ethically and to a high standard.
Ultimately, choosing Latin America for an FIH trial is more than a tactical cost-saving measure. It can be a pivotal strategic decision that accelerates the entire development program. Generating crucial human data faster and more efficiently can de-risk the technology, attract subsequent rounds of investment, and significantly shorten the overall path to global market approval.
Frequently Asked Questions (FAQ)
Q1: What if my device uses a novel material with no existing data?A: In this specific scenario, a justification-only approach based on existing data is unlikely to be sufficient. You will be required to generate new data to establish a safety profile. The ISO 10993 framework will be your guide. The process would start with extensive physicochemical characterization and a battery of in vitro tests. If, after these steps, significant biological risks remain that can only be addressed by an in vivo model, then targeted, well-justified animal studies may become necessary. The crucial point is that these studies would be a last resort to answer a specific question, not a default first step.
Q2: Is a toxicologist's sign-off mandatory for my justification?A: While a line-item on an application form may not explicitly say "Toxicologist Signature Required," it is practically mandatory for a successful submission. Regulatory bodies across the region expect an "expert interpretation" of the safety data. A preclinical package that contains complex chemical characterization data but lacks a credible toxicological risk assessment signed by a qualified expert is highly likely to be considered incomplete, leading to rejection or significant delays while the agency issues deficiency letters.
Q3: How does this approach compare to FDA or EU MDR preclinical requirements?A: The fundamental scientific principles are the same. The U.S. FDA and European regulations (MDR) also operate on a risk-based approach guided by ISO 10993 and ISO 14971. The core evidence needed to prove safety is universal. The key difference often lies in the implementation, regulatory agility, and risk tolerance for FIH trials. Latin American agencies can sometimes be more pragmatic and interactive in their review process for early-stage trials, which contributes to the faster approval timelines. However, the high-quality scientific evidence you generate for a Latin American submission is the same evidence you would need to prepare for the FDA or a European Notified Body.
Q4: Can I use my existing FDA preclinical data package for a submission in Latin America?A: Yes, absolutely. If your preclinical data package was prepared according to modern standards (i.e., reflecting the post-2018 ISO 10993-1 principles, with robust chemical characterization and a toxicological risk assessment), it is very likely to be sufficient to meet the scientific requirements in Latin America. The primary work will involve re-packaging this evidence into a comprehensive justification memorandum, ensuring it directly addresses the biological risks of your device, and translating key summary documents and application materials into Spanish or Portuguese as required by the specific country.
Q5: What exactly is a "New Approach Methodology" (NAM)?A: A New Approach Methodology (NAM) is a broad term for any non-animal technology, methodology, or approach used to assess the safety of a device or its component materials. This includes a wide range of techniques such as advanced in vitro methods (e.g., "organ-on-a-chip" technology, high-throughput screening), in silico methods (computer modeling and quantitative structure-activity relationship analysis), and the combination of advanced analytical chemistry with toxicological modeling. Brazil's ANVISA is a regional leader in formally recognizing and encouraging the use of NAMs in regulatory submissions.
List of Sources
Navigating Preclinical Requirements for First-in-H.pdf
Breaking the unvirtuous cycle: barriers and opportunities for research and development in Paraguay. A case study - PubMed Central, accessed August 12, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC10687402/