
For medical device sponsors, the path to First-in-Human (FIH) data has fundamentally shifted.
The traditional routes for Early Feasibility Studies (EFS)—the US and Western Europe—are facing unprecedented congestion. With the EU Medical Device Regulation (MDR) creating 18-month backlogs for clinical investigation approvals and the US FDA demanding increasingly mature GLP (Good Laboratory Practice) data even for pilot studies, innovation is stalling.
In 2025, smart capital is pivoting. Emerging jurisdictions like Brazil, Mexico, Chile, Argentina, and Serbia are offering a strategic alternative: the ability to generate robust, regulatory-grade safety data 6–9 months faster than traditional markets.
This guide decodes the regulatory, legal, and importation frameworks of these five key jurisdictions, helping sponsors of Class III and novel devices navigate the "Tier 2" landscape.
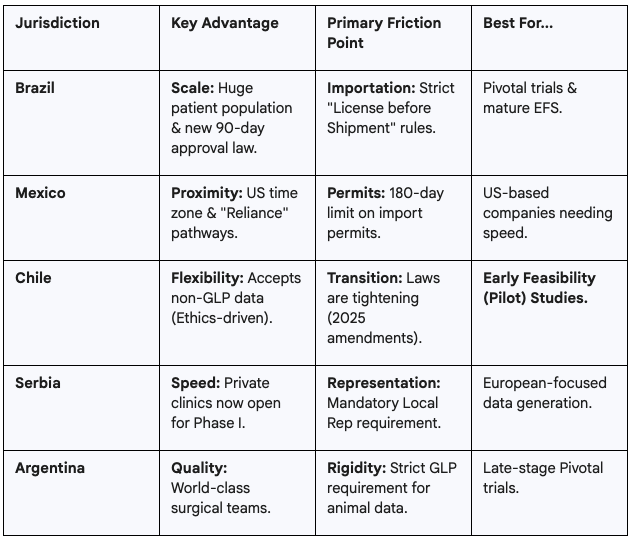
Brazil has undergone a legislative revolution, shedding its reputation for bureaucracy to become a top-tier destination for clinical research.
Enacted in May 2024, this law dismantled the notorious "double approval" bottleneck. Previously, studies required sequential approval from both local and national ethics bodies.
Importation remains the most complex operational hurdle.
Mexico offers proximity to the US and a rapidly modernizing regulatory agency, COFEPRIS.
Effective September 1, 2025, COFEPRIS implemented a new Abbreviated Regulatory Pathway. While primarily for market registration, this cultural shift impacts clinical trials. Protocols for devices that have received feedback or "Study Risk Determinations" from the US FDA are now viewed with greater deference, speeding up authorization.
To bypass internal backlogs, sponsors should utilize Authorized Pre-Assessment Units (UHAPs).
Unlike other regions, Mexico’s Permiso Sanitario de Importación is quantity-specific and typically valid for only 180 days. Supply chains must be tightly coordinated; if recruitment lags, the permit may expire, halting shipments.
Chile is arguably the most favorable jurisdiction for early-stage startups possessing high-quality but non-GLP pre-clinical data.
Unlike Brazil or Mexico, Chile’s Public Health Institute (ISP) often delegates the primary review of early-phase trials to accredited Scientific Ethics Committees (CECs).
To move devices into Chile, sponsors use the GICONA system to request a Certificado de Destinación Aduanera (CDA). This digital process is highly efficient, often clearing goods in under 10 days.
Serbia is not in the EU, but its data is EU-accepted. It serves as a high-speed bridge for companies targeting eventual CE Marking.
In December 2024, Serbia amended its Clinical Trials Rulebook to allow private healthcare institutions to conduct Phase I and Early Feasibility studies.
Foreign sponsors cannot operate directly. You must appoint a Local Legal Representative to submit the application to the regulatory agency (ALIMS). Note that Serbia enforces GDPR-equivalent data privacy laws; a separate Data Protection Representative may also be required.
Argentina offers world-class clinicians but maintains a rigid regulatory stance regarding pre-clinical testing.
Argentina’s agency, ANMAT, is a Level 4 Reference Authority. Under Disposition 6677/10, ANMAT strictly requires that pre-clinical studies supporting human trials be conducted under certified Good Laboratory Practice (GLP).
A critical 2025 update (Disposition 8799/2025) mandates that imported medical devices must have at least six months of remaining shelf life upon entry. For early-stage prototypes with limited stability data, this can be a major logistical or compliance risk.
For a standard Early Feasibility Study (10–15 patients) involving a novel Class III device: