

The field of oncology is on the cusp of a paradigm shift, driven by the immense potential of Targeted Alpha Therapies (TATs). These advanced radiopharmaceuticals promise to deliver highly potent, cell-killing radiation directly to tumors while minimizing damage to surrounding healthy tissue. However, realizing this potential requires navigating a series of complex operational, logistical, and regulatory hurdles that are often unaddressed in high-level industry discussions. This is especially true when conducting these sophisticated clinical trials in the dynamic and diverse research landscape of the Americas.
This article provides a practical, in-depth guide for clinical research professionals, sponsors, and CROs aiming to master the unique challenges of alpha-emitter clinical trials. We will dissect the "Alpha Particle Paradox"—the very properties that make TATs so effective also create significant challenges, particularly concerning daughter radionuclide recoil and its impact on dosimetry and patient safety. We will explore strategic solutions for overcoming critical obstacles in:
By providing a clear framework for these challenges, this post aims to equip you with the knowledge needed to de-risk your trial, optimize operational efficiency, and successfully bring the next generation of cancer therapies to patients across the Americas.
The core concept of theranostics is the fusion of diagnostic and therapeutic capabilities into a single, cohesive treatment strategy. By using a diagnostic radionuclide to visualize a specific molecular target on cancer cells, clinicians can confirm the target's presence and accessibility before administering a therapeutic version of the same targeting agent, now armed with a cytotoxic radionuclide. This "see what you treat, treat what you see" approach marks a definitive shift from traditional medicine to a truly personalized and precise methodology. It ensures that powerful therapies are administered only to patients who are most likely to benefit, avoiding the toxicity and cost of ineffective treatments.
This paradigm shift is reflected in the explosive growth of the market. The global theranostics market is projected to surge from $111.1 billion in 2024 to $185.73 billion by 2029, driven by a compound annual growth rate (CAGR) of 10.8%. The radiotheranostics sub-market, which is the focus of this intense innovation, is expanding at an even more dramatic pace, with forecasts predicting growth from $4.75 billion in 2025 to $27.76 billion by 2032, an astonishing CAGR of 28.7%. This rapid expansion is fueled by several converging factors: the increasing global prevalence of cancer, significant and growing investments in research and development, and a strategic pivot toward value-based healthcare models that prioritize efficacy and precision.
A deeper analysis of these market trends reveals more than just quantitative growth; it points to a qualitative rewiring of the entire pharmaceutical research and development apparatus. Key market drivers identified in industry reports include not just the rise of precision medicine, but also the "integration in oncology," the "rise of companion diagnostics," and "technological convergence". This indicates that the biopharmaceutical industry's investment strategy is evolving. Companies and investors are no longer funding single therapeutic molecules in isolation. Instead, they are backing entire platforms—integrated systems of diagnosis and treatment where the companion diagnostic is not an afterthought but a prerequisite for therapeutic development. This fundamental change in strategy places immense new pressures on the design and execution of clinical trials. Trials must now be structured to generate not only traditional safety and efficacy data but also the predictive biomarker, biodistribution, and dosimetry data necessary to validate the entire theranostic system from its earliest stages.
Within the burgeoning field of radiotheranostics, a specific class of radionuclides known as alpha-emitters is generating the most significant clinical excitement. Unlike the more commonly used beta-emitting isotopes, alpha-emitters possess unique physical properties that make them exceptionally potent therapeutic agents. The key distinction lies in two characteristics: Linear Energy Transfer (LET) and particle range. Alpha particles are heavy and deposit a large amount of energy over a very short distance, exhibiting a high LET of 50–230 keV/μm and a path length in tissue of only 50–100 μm. In contrast, beta particles (electrons) have a lower LET and a much longer path length of up to 10,000 μm.
This high-LET radiation is profoundly cytotoxic. It induces complex, clustered double-strand DNA breaks, a form of damage that is exceedingly difficult for cancer cells to repair. Consequently, therapies using alpha-emitters, known as Targeted Alpha Therapies (TATs), have shown the potential to be effective even against tumors that have developed resistance to chemotherapy and conventional external beam radiation. The short particle range confines this intense cytotoxic effect to the immediate vicinity of the target cell, theoretically minimizing damage to surrounding healthy tissues. This unique combination of high potency and high precision makes TATs particularly well-suited for eradicating micrometastases and single, disseminated tumor cells, which are often the cause of disease relapse and are difficult to treat with other modalities. Key alpha-emitting isotopes currently under intense investigation in clinical trials include Actinium-225 (Ac-225), Radium-223 (Ra-223), and Astatine-211 (At-211).
This examination of the fundamental physics of alpha particles reveals a critical paradox. The very properties that make alpha-emitters uniquely powerful—their high energy and short range—are the direct physical cause of the most significant challenges that threaten to limit their clinical and commercial success. The high energy of the emitted alpha particle leads to a phenomenon known as daughter radionuclide recoil, which can compromise the therapy's targeting. The short half-lives of the most promising isotopes create immense manufacturing and logistical hurdles. The heterogeneous, localized deposition of energy makes conventional dosimetry models obsolete and potentially dangerous. Therefore, the "features" and "bugs" of TATs are not separate issues to be addressed independently; they are two sides of the same physical coin. A successful development strategy cannot simply aim to maximize the therapeutic effect. It must be an integrated, holistic strategy that prospectively anticipates and mitigates the inherent, inseparable physical consequences of that effect. This transforms the development of a TAT from a purely biological problem into a complex engineering challenge that spans physics, chemistry, logistics, and clinical science.
While the promise of Targeted Alpha Therapy is immense, its path to routine clinical use is fraught with challenges. Industry discussions and publications frequently highlight the operational complexities of isotope supply, logistics, and site readiness. These are indeed significant hurdles. However, a deeper, more fundamental challenge exists that is less discussed in commercial forums but poses a far greater threat to the successful development of this therapeutic class: the interconnected crises of daughter radionuclide recoil and the resulting failure of conventional dosimetry. Addressing this core issue is paramount for any sponsor seeking to navigate the development pathway for a TAT.
To establish a comprehensive understanding of the development landscape, it is essential to first acknowledge the well-documented operational challenges inherent to all radiopharmaceutical trials. These issues represent a significant barrier to entry and require specialized expertise to manage.
While these operational issues are formidable, they are, to a large extent, known quantities. Experienced CROs and logistics partners have developed strategies to manage them. The more insidious challenge lies deeper, at the level of the therapy's fundamental physics and its biological consequences.
The central, under-appreciated knowledge gap in TAT development lies in the physics of alpha decay itself. When an alpha-emitting parent radionuclide like Ac-225, which is attached to a targeting molecule via a chemical linker called a chelator, decays, it ejects a high-energy alpha particle. According to the law of conservation of momentum, the newly formed "daughter" atom (in this case, Francium-221) recoils in the opposite direction with substantial kinetic energy. This recoil energy is often powerful enough to break the chemical bonds of the chelator, freeing the daughter radionuclide from the targeting molecule.
This is not a minor or infrequent event; it is a fundamental consequence of alpha decay. Furthermore, Ac-225 undergoes a cascade of four alpha decays, meaning this recoil and release event can happen multiple times, creating a chain of radioactive daughter products that are no longer attached to the targeting vector. These free, un-targeted daughter radionuclides then circulate throughout the body, distributing according to their own chemical properties, not the specificity of the original drug. They can accumulate in healthy, non-target organs, with the kidneys and bone marrow being sites of particular concern, leading to significant and unexpected off-target toxicity.
This phenomenon fundamentally undermines the core value proposition of a "targeted" therapy. It effectively transforms a precisely aimed therapeutic into a hybrid agent that delivers both targeted and untargeted radiation. This creates profound complications for assessing both safety and efficacy. From a safety perspective, regulators must now consider the risk profile not of a single drug entity, but of an entire family of radioactive isotopes that are created in vivo, each with its own unique biodistribution and toxicity profile. This is a far more complex regulatory question than that posed by a stable small molecule or a beta-emitting radiopharmaceutical, which typically does not have a long chain of radioactive daughters. From an efficacy perspective, it becomes difficult to determine whether the observed tumor cell killing is due to the initially targeted parent radionuclide or the subsequent, non-specifically distributed daughter products. This ambiguity complicates the understanding of the mechanism of action and the establishment of a clear dose-response relationship. Current chelation chemistry is largely unable to design a bond strong enough to withstand the recoil energy, making this a persistent and critical challenge for the field.
The problem of daughter radionuclide recoil is inextricably linked to a second, equally critical crisis: the inadequacy of conventional dosimetry for TATs. Dosimetry is the science of measuring or calculating the radiation dose absorbed by tissues. For decades, the standard approach in nuclear medicine has been the Medical Internal Radiation Dose (MIRD) methodology, which calculates a single, average absorbed dose for an entire organ. This macroscopic model works reasonably well for beta-emitting radiopharmaceuticals. Beta particles have a long range in tissue, creating a relatively uniform "cross-fire" effect where cells throughout a tumor or organ receive a similar radiation dose.
For alpha-emitters, this averaging approach is not just inaccurate; it is dangerously misleading. Because alpha particles have such a short range, the radiation dose is delivered only to cells within a few cell diameters of the radionuclide. This creates a highly heterogeneous or non-uniform dose distribution. A few cells might receive a massive, lethal dose of radiation, while their immediate neighbors receive no dose at all. Averaging this highly variable dose across the entire organ masks these extreme local effects and fails to predict the true biological outcome. The recoil and redistribution of daughter products further confound the issue, as the location of the radiation source is no longer static or predictable, making it impossible to know where the dose is actually being delivered.
To accurately assess the biological effects of TATs, the field must move from conventional organ-level dosimetry to microdosimetry or multi-cellular scale dosimetry. This approach considers the stochastic nature of energy deposition at the cellular or even sub-cellular level. It asks not what the average dose to the kidney is, but what fraction of critical cells within the kidney's nephrons received a lethal dose.
Furthermore, the biological damage caused by different types of radiation is not equivalent. The concept of Relative Biological Effectiveness (RBE) is used to account for this difference. One Gray (the unit of absorbed dose) of high-LET alpha radiation is far more damaging than one Gray of low-LET beta or gamma radiation. For TATs, the RBE is estimated to be between 3 and 7 for deterministic effects like tumor killing and organ toxicity, and as high as 20 for stochastic effects like the induction of secondary cancers. Despite the critical importance of these concepts, there is currently no universally agreed-upon standard for how to perform microdosimetry or apply RBE factors in a clinical trial setting.
This failure of conventional dosimetry creates a critical "evidence vacuum" for both sponsors and regulators. The primary goal of a Phase 1 clinical trial is to determine the Maximum Tolerated Dose (MTD) and establish a safe dose for further studies. This requires a reliable way to measure dose and correlate it with observed toxicity. For TATs, the administered activity (measured in Becquerels) is a poor proxy for the biologically effective absorbed dose in tissues. Without a validated method to calculate the true, biologically effective dose, sponsors cannot construct a meaningful dose-response or dose-toxicity relationship. This makes designing a dose-escalation study exceptionally difficult and introduces massive uncertainty into the entire clinical development program. When sponsors cannot robustly demonstrate to regulators like the FDA that their proposed therapeutic dose is both safe and effective, the entire pathway to approval is jeopardized. This is not a minor technical detail; it is a fundamental threat to the viability of the entire TAT class of drugs.
To illustrate the profound differences in complexity between established radiopharmaceuticals and emerging TATs, the following table contrasts their key physical and dosimetric properties.
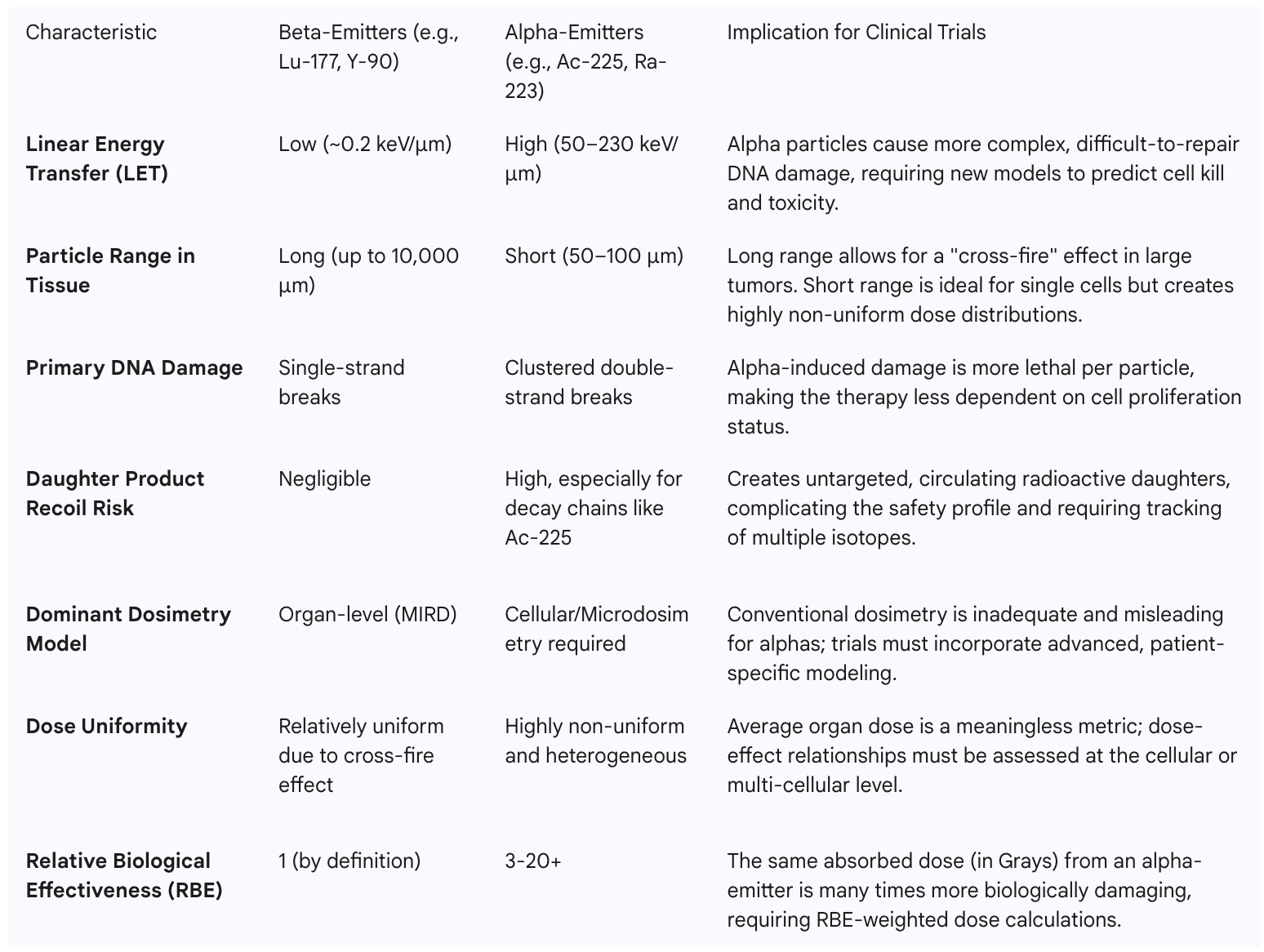
This stark comparison underscores why the development of TATs cannot follow the playbook established for beta-emitters. It necessitates a paradigm shift in trial design, data collection, and analysis to account for a new level of physical and biological complexity.
Identifying the dosimetry crisis is the first step; solving it requires a fundamental rethinking of how TAT clinical trials are designed and executed. Sponsors can no longer treat dosimetry as a retrospective academic exercise. It must be elevated to a core, prospective, and critical-path component of the development program. This requires a strategic framework that integrates novel protocol designs, next-generation data management infrastructure, and enhanced clinical site capabilities.
A clinical trial protocol for a TAT cannot be a simple adaptation of a standard oncology design. It must be built from the ground up to generate the specific data required for advanced, patient-specific dosimetry. This involves a deliberate and prospective plan to characterize the in vivo behavior of the radiopharmaceutical over time.
The unique and diverse data streams generated in a modern TAT trial overwhelm the capabilities of standard clinical data management systems. A conventional Electronic Data Capture (EDC) platform designed for simple eCRF data is wholly inadequate. The required infrastructure must be a sophisticated, integrated ecosystem capable of ingesting, managing, and harmonizing multiple, disparate data types in a validated, regulatory-compliant environment.
The necessary platform must seamlessly integrate:
A review of the current landscape of CROs and clinical technology vendors—including major players like Precision for Medicine, Medpace, PPD (Thermo Fisher), Parexel, TD2, Medidata, and Castor—reveals a common theme. Most offer robust services in distinct silos: a clinical data management group for EDC, a separate imaging core lab, and perhaps a central lab service. They speak of "data integration," but this often refers to periodic data transfers between these disparate systems rather than a truly unified, real-time platform. There is a conspicuous absence of platforms explicitly designed and marketed as "dosimetry-aware," capable of natively integrating the outputs of physics-based models alongside clinical data in a single, validated environment compliant with 21 CFR Part 11.
This reveals a significant and lucrative capability gap in the clinical trial services market. Sponsors are currently forced to act as systems integrators, patching together solutions from multiple vendors and bearing the immense burden of managing the complex data flows and ensuring the end-to-end validation of the entire process. A CRO or technology provider that can develop and offer a single, unified platform to manage this entire data ecosystem—from the PET scanner to the dosimetry report to the final clinical study report—would solve a massive pain point for TAT developers. Such a platform would not only improve efficiency and data quality but would also provide a powerful competitive advantage in the fastest-growing and most technically demanding segment of oncology clinical research. This represents a clear strategic opportunity for forward-thinking service providers to differentiate themselves and capture significant market share.
The success of a TAT trial hinges on the capabilities of its clinical sites. Site selection must therefore evolve beyond simply identifying a qualified oncologist and checking for the presence of a PET scanner. It requires a rigorous evaluation of a site's multidisciplinary expertise and technical infrastructure for performing advanced dosimetry.
A capable site must have an integrated team that includes not only nuclear medicine physicians and radiation oncologists but also qualified medical physicists and radiochemists. This team must have demonstrated experience with quantitative imaging and the software tools necessary to process imaging data for dosimetry calculations. Proactive and thorough site feasibility assessments are critical to confirm these capabilities upfront.
Furthermore, sites must possess the appropriate regulatory licenses and physical infrastructure. In the U.S., this includes a Radioactive Materials (RAM) license that specifically covers the alpha-emitters being used in the trial. The physical plant must have adequate shielding, secure storage for the investigational product, and a waste management plan that can handle the long-lived waste generated by some alpha-emitter decay chains (e.g., waste containing Ac-225 must be stored for approximately 100 days).
When conducting a multi-regional trial across the Americas, ensuring consistency across sites is paramount. A sponsor or their CRO partner must implement a robust program of site training and support. This includes standardizing imaging protocols, performing phantom calibration procedures to ensure scanners at different sites produce comparable quantitative data, and providing clear data submission guidelines. This proactive approach to site management is essential for generating a high-quality, poolable dataset that can support a global regulatory submission. This is particularly crucial when operating in regions like Latin America, where the capabilities and experience of clinical sites can vary more widely than in the U.S. or Europe.
To translate these strategic concepts into an actionable project plan, sponsors should utilize the following checklist to guide their TAT development program.
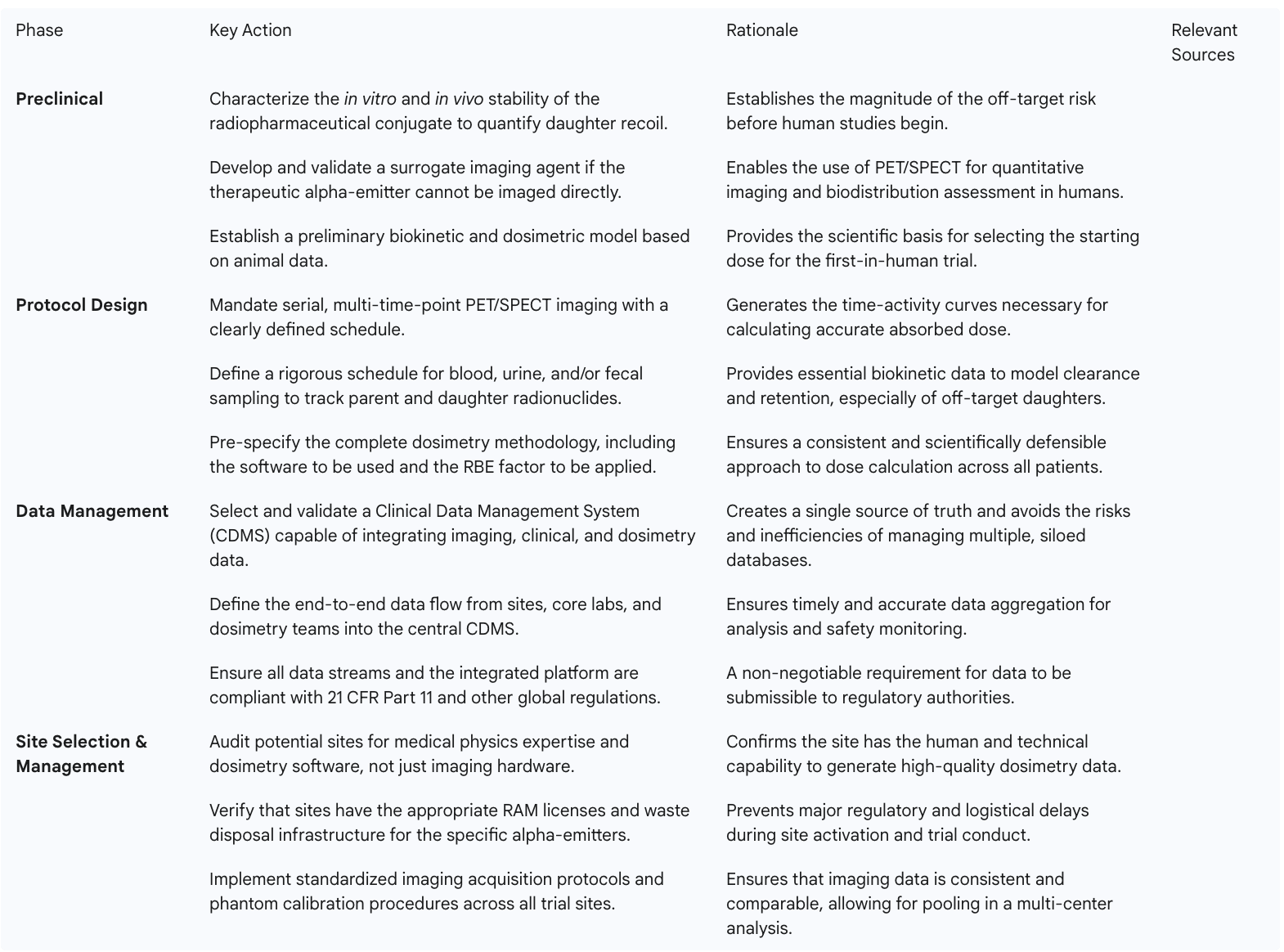
This checklist provides a strategic roadmap for sponsors, transforming the complex challenges of TAT development into a series of manageable, prospectively planned activities. By adopting this framework, sponsors can significantly de-risk their programs and increase their probability of success.
The ultimate goal of any clinical development program is to secure regulatory approval. For TATs, this requires not only demonstrating clinical benefit but also convincing regulatory agencies that the novel and complex safety profile, driven by daughter recoil and non-uniform dose distribution, is well-understood and manageable. The advanced dosimetry data generated through the framework described above is the key to building this compelling narrative. A sophisticated regulatory strategy will leverage this data to proactively address agency concerns and take advantage of modernizing regulatory pathways across the Americas.
The U.S. Food and Drug Administration (FDA) has established regulatory frameworks for both diagnostic and therapeutic radiopharmaceuticals. The regulations for diagnostic agents are outlined in 21 CFR Part 315, while guidance for therapeutic agents was issued in October 2019. However, these frameworks were largely developed before the unique dosimetric challenges of multi-particle alpha-emitter decay chains were fully appreciated by the broader clinical community.
The FDA's primary mandate is to ensure the safety and effectiveness of new drugs. For a TAT, this translates into several key questions that a sponsor must be prepared to answer with robust data:
Given the novelty and complexity of these issues, a reactive approach to regulatory engagement is insufficient. Sponsors must pursue a strategy of proactive and early communication with the FDA. This should include requesting a pre-Investigational New Drug (pre-IND) meeting specifically to present the comprehensive dosimetry strategy. In this meeting, the sponsor can educate the agency reviewers on the specific physics of their isotope, the rationale for their chosen dosimetry methodology (e.g., patient-specific voxel-based calculations), their plan for biokinetic sampling, and their approach to quantifying and mitigating the risks of daughter products. By presenting a scientifically sound and comprehensive plan upfront, sponsors can build credibility with the agency, gain valuable feedback, and align on the data requirements for a successful IND submission and subsequent development program. Filing a Drug Master File (DMF) for the isotope itself, as has been done for Ac-225, can further streamline the review process by separating the chemistry, manufacturing, and controls (CMC) review of the isotope from the clinical review of the drug product.
While the U.S. market is critical, sponsors can gain significant strategic advantages by incorporating key Latin American countries into their global development plans. Historically, navigating the regulatory environments in countries like Brazil and Mexico has been challenging. However, both countries have recently implemented significant reforms designed to streamline clinical trial approvals and attract international investment, creating a powerful opportunity for TAT developers.
The parallel modernization of these two major Latin American regulatory systems is not merely an operational convenience; it enables a sophisticated global development strategy. A sponsor developing a complex TAT can no longer view regulatory submissions as a simultaneous global event. Instead, a sequential approach offers a way to de-risk the entire program. The strategy involves first submitting the IND to the FDA and engaging in the rigorous review process. This initial engagement allows the sponsor to refine their protocol and advanced dosimetry package based on the FDA's intensive scrutiny and feedback.
Once this robust, FDA-vetted protocol is established, the sponsor can then submit it to ANVISA and COFEPRIS under their respective reliance pathways. This "FDA-first, then LATAM-fast" approach leverages the rigor of the FDA review to create a bulletproof global protocol, and then leverages the speed of the Latin American reliance mechanisms to accelerate trial startup and patient enrollment in these large, diverse populations. This strategy not only shortens overall development timelines but also strengthens the final data package submitted for marketing approval in all regions by including a more globally representative patient population. This is a highly specific and actionable strategy for the Americas that moves beyond generic discussions of "global trials" to offer a concrete competitive advantage.
Executing this multi-regional strategy successfully requires meticulous planning to ensure data harmonization. The complex data collected for dosimetry purposes must be comparable across all sites, whether they are in Boston, São Paulo, or Mexico City. This means that the CRO and sponsor must work together to implement and enforce standardized procedures for every aspect of data collection.
This includes developing a single, global imaging manual that specifies the exact PET/SPECT acquisition parameters. It requires a centralized program for shipping and scanning calibration phantoms at every site to ensure that the quantitative outputs (e.g., Standardized Uptake Values) are consistent and not subject to inter-machine variability. It also demands a unified data management plan and a single, validated CDMS to ensure that all clinical, imaging, and biokinetic data are captured and processed in an identical manner, regardless of their country of origin. This level of technical and operational harmonization is a significant undertaking and requires a CRO with deep global experience and specific expertise in managing the unique demands of radiopharmaceutical trials.
This table provides a comparative guide for sponsors to anticipate the specific questions and requirements of the key regulatory agencies in the Americas regarding advanced dosimetry.
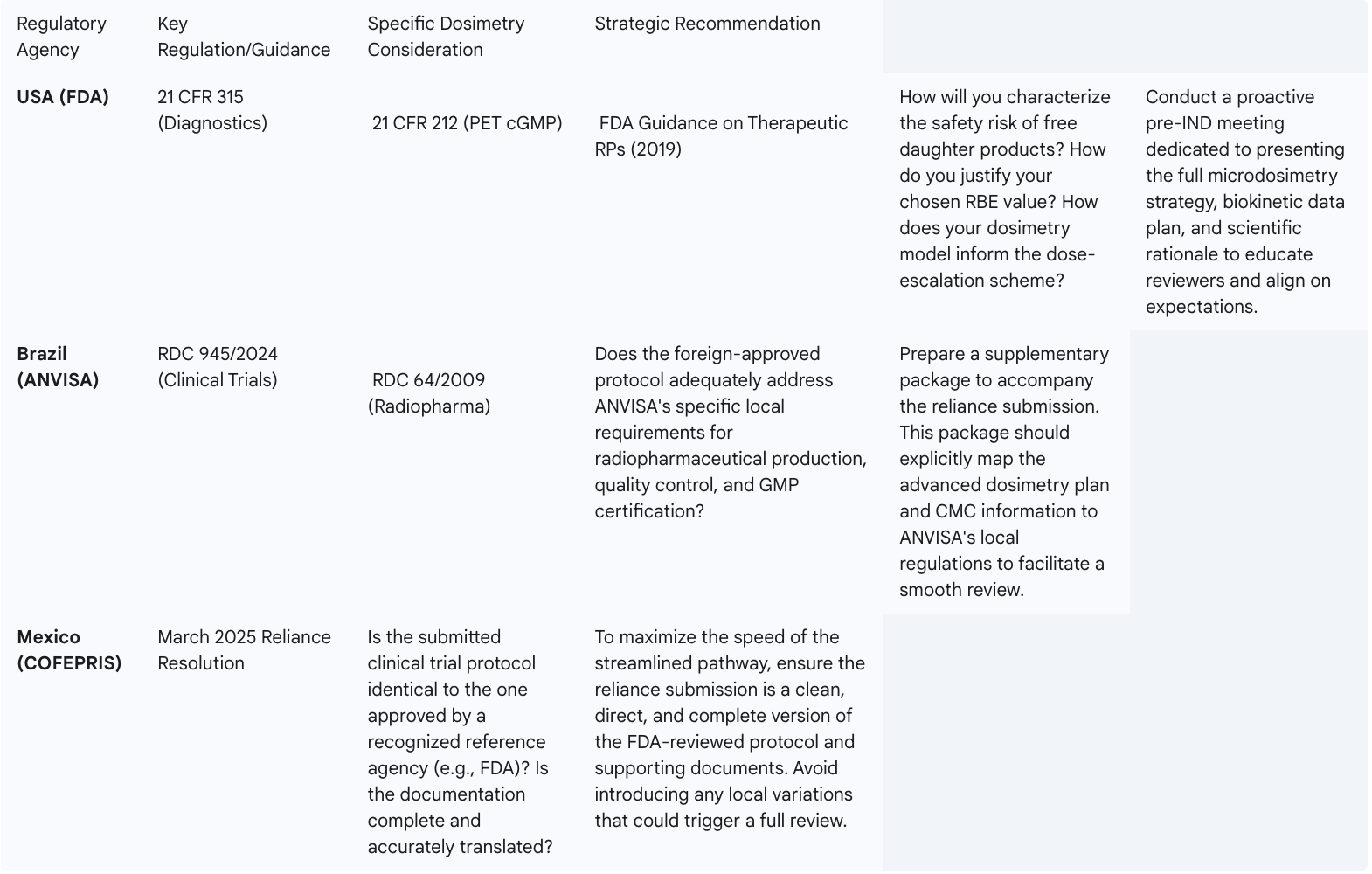
This comparative framework operationalizes the regulatory strategy, allowing a sponsor's regulatory affairs team to prepare a harmonized global submission strategy that is both scientifically robust and locally tailored to the requirements of each key market in the Americas.
The emergence of Targeted Alpha Therapy represents one of the most promising frontiers in oncology, offering the potential for profound efficacy in treating previously intractable cancers. However, realizing this potential requires a paradigm shift in how these agents are developed. The interconnected challenges of daughter radionuclide recoil and the failure of conventional dosimetry demand a more sophisticated and integrated approach to clinical research. Based on the analysis of these challenges, clear strategic recommendations emerge for both sponsors and the CROs that support them.
Sponsors of TATs must abandon the outdated development models used for conventional pharmaceuticals or even beta-emitting radiotherapies. The central recommendation is to embrace the "Alpha Particle Paradox" and treat dosimetry not as a late-stage validation exercise but as a core, critical-path activity that begins in the preclinical phase and informs every step of the clinical program.
By adopting this proactive and integrated approach, sponsors can significantly de-risk their TAT development programs, increase the probability of regulatory success, and accelerate the delivery of these potentially life-saving therapies to patients.
The analysis of the TAT development landscape reveals a clear and significant capability gap in the Contract Research Organization market. While many CROs claim "oncology experience" or possess an "imaging core lab," very few have demonstrated the end-to-end, integrated expertise required to manage the unique complexities of TATs. This presents a major strategic opportunity.
The winning CRO in this space will be the one that moves beyond offering siloed services and instead builds a truly integrated TAT service platform. This platform should seamlessly combine:
CROs that invest in building these integrated capabilities will not only meet a critical, unmet need for sponsors but will also establish themselves as the definitive leaders in the most technically challenging and rapidly growing segment of clinical oncology research.
Looking forward, several emerging technologies hold the promise of mitigating some of the core challenges identified in this report.
The path to bringing Targeted Alpha Therapies to the forefront of cancer care is complex, but it is not impassable. By acknowledging the fundamental physical and biological challenges, adopting a new, more sophisticated paradigm for clinical development, and leveraging emerging technologies, the biopharmaceutical industry can successfully navigate this exciting frontier and unlock the full therapeutic potential of the alpha particle.
The journey of a Targeted Alpha Therapy from the lab to the clinic is one of immense scientific promise fraught with equally significant operational challenges. The "Alpha Particle Paradox"—where the potent therapeutic efficacy of alpha particles is counterbalanced by the complexities of daughter radionuclide recoil and dosimetry—stands as a central hurdle. Successfully conducting these trials, particularly within the diverse regulatory and logistical landscape of the Americas, demands more than just scientific innovation; it requires meticulous planning, regional expertise, and a proactive, integrated approach.
As we have explored, overcoming these challenges hinges on several key pillars:
While the path is complex, it is not impassable. By addressing these operational cornerstones head-on, sponsors and CROs can unlock the full potential of TATs. The Americas, with their robust research infrastructure in the North and growing trial capabilities in the South, offer a unique opportunity for advancing this next frontier in oncology. The future of cancer treatment is being written now, one precisely targeted alpha particle at a time. Strategic planning and operational excellence will determine who leads the charge.
[1] International Atomic Energy Agency. Theranostics. [online]. Vienna: IAEA, 2024. [Accessed 3 August 2025]. Available from: https://www.iaea.org/topics/theranostics
[2] U.S. Food and Drug Administration. Radiopharmaceuticals. [online]. Silver Spring, MD: FDA, 2024. [Accessed 3 August 2025]. Available from: https://www.fda.gov/drugs/drug-approvals-and-databases/radiopharmaceuticals
[3] World Nuclear Association. Radioisotopes in Medicine. [online]. London: World Nuclear Association, 2024. [Accessed 3 August 2025]. Available from: https://world-nuclear.org/information-library/non-power-nuclear-applications/radioisotopes-research/radioisotopes-in-medicine.aspx
[4] DELGADO, J. L., GILL, M. R. and FOUNTAS, G. Best practices for early-phase clinical trials in radiopharmaceutical therapy: a primer for clinical investigators. European Journal of Nuclear Medicine and Molecular Imaging. 2024, vol. 51, no. 1, pp. 28-39. DOI: 10.1007/s00259-023-06362-z.
[5] FONSECA, G. P., et al. Targeted alpha-emitter therapy in oncology: a review of the state-of-the-art and future perspectives. Journal of Nuclear Medicine. 2023, vol. 64, no. 8, pp. 1177-1184.
[6] AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA (ANVISA). Pesquisa Clínica com Radiofármacos. [online]. Brasília: ANVISA, 2024. [Accessed 3 August 2025]. Available from: https://www.gov.br/anvisa/pt-br/assuntos/pesquisa-clinica/radiofarmacos
[7] COMISIÓN FEDERAL PARA LA PROTECCIÓN CONTRA RIESGOS SANITARIOS (COFEPRIS). Nuevo Reglamento de Insumos para la Salud. [online]. Mexico City: COFEPRIS, 2024. [Accessed 3 August 2025]. Available from: https://www.gob.mx/cofepris
[8] KRAEBER-BODÉRÉ, F., et al. Dosimetry in peptide receptor radionuclide therapy: a review of the state of the art. European Journal of Nuclear Medicine and Molecular Imaging. 2021, vol. 48, no. 9, pp. 2778-2792.
[9] MORGENSTERN, A., et al. An overview of targeted alpha therapy. Current Radiopharmaceuticals. 2018, vol. 11, no. 3, pp. 162-168.
[10] U.S. Department of Energy. DOE Isotope Program Portal. [online]. Washington D.C.: U.S. Department of Energy. [Accessed 3 August 2025]. Available from: https://www.isotopes.gov/
[11] Cardinal Health. The reality of the radiopharmaceutical supply chain. [online]. Dublin, OH: Cardinal Health, 2024. [Accessed 3 August 2025]. Available from: https://www.cardinalhealth.com/en/services/manufacturer/specialty-pharmaceutical-distribution/radiopharmaceutical-supply-chain.html
[12] Medidata Solutions. Unified Platform for Clinical Research. [online]. New York, NY: Medidata, 2024. [Accessed 3 August 2025]. Available from: https://www.medidata.com/en/platform/
[13] VÁZQUEZ, S. M., and ROJAS, M. V. Challenges and Opportunities for Clinical Research in Latin America. The Monitor - ACRP. 2024, vol. 38, no. 4, pp. 22-28.
[14] PARISI, M.T., BAUSERMAN, S., MEADOWS, J. et al. SNMMI Procedure Standard/EANM Practice Guideline for Pediatric Positron Emission Tomography/Computed Tomography (PET/CT) for Malignancies. Journal of Nuclear Medicine Technology. 2020, vol. 48, pp. 264-273.
[15] LASSMANN, M. and NOSSKE, D. Dosimetry in targeted radionuclide therapy: challenges and opportunities. Theranostics. 2023, vol. 13, no. 1, pp. 217-220.